Code
── Attaching packages ─────────────────────────────────────── tidyverse 1.3.1 ──
✔ ggplot2 3.4.2 ✔ purrr 1.0.1
✔ tibble 3.2.1 ✔ dplyr 1.1.2
✔ tidyr 1.3.0 ✔ stringr 1.5.0
✔ readr 2.1.4 ✔ forcats 1.0.0
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
Code
# Get the most recent set of reviews <- dir (here:: here ("outputs" , "reviews" ), "reviews.*rds" , full.names = TRUE ) %>% sort (decreasing = TRUE ) %>% first () %>% readRDS ()# And most recent studies-by-taxonomy <- readRDS (here:: here ("outputs" , "taxa" , "study_taxa.rds" ))
How many papers have we scored?
Code
<- reviews %>% mutate (across (everything (), \(x) ifelse (x == "NA" , NA , x))) %>% filter (reviewed)nrow (scored_papers)
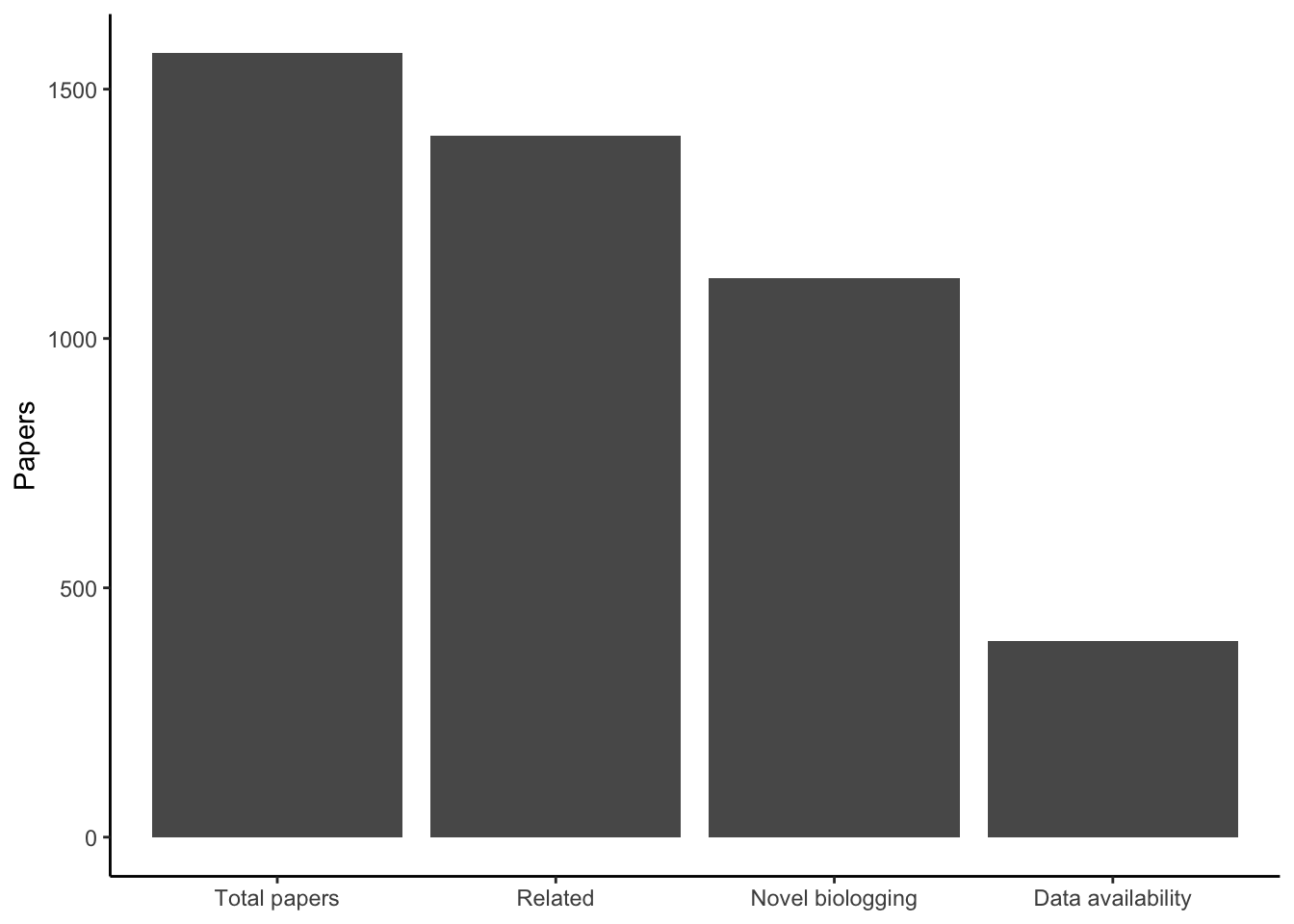
Breakdown of papers by relatedness, novel biologging, and data availability statement.
Code
%>% summarize (` Total papers ` = n (),` Related ` = sum (manuscript_type != "U" , na.rm = TRUE ),` Novel biologging ` = sum (novel_biologging == "Y" ,na.rm = TRUE ),` Data availability ` = sum (biologging_availability == "Y" ,na.rm = TRUE )) %>% pivot_longer (everything ()) %>% mutate (name = fct_reorder (name, value, .fun = \(x) - x)) %>% ggplot (aes (name, value)) + geom_col () + labs (y = "Papers" ) + theme_classic () + theme (axis.title.x = element_blank ())
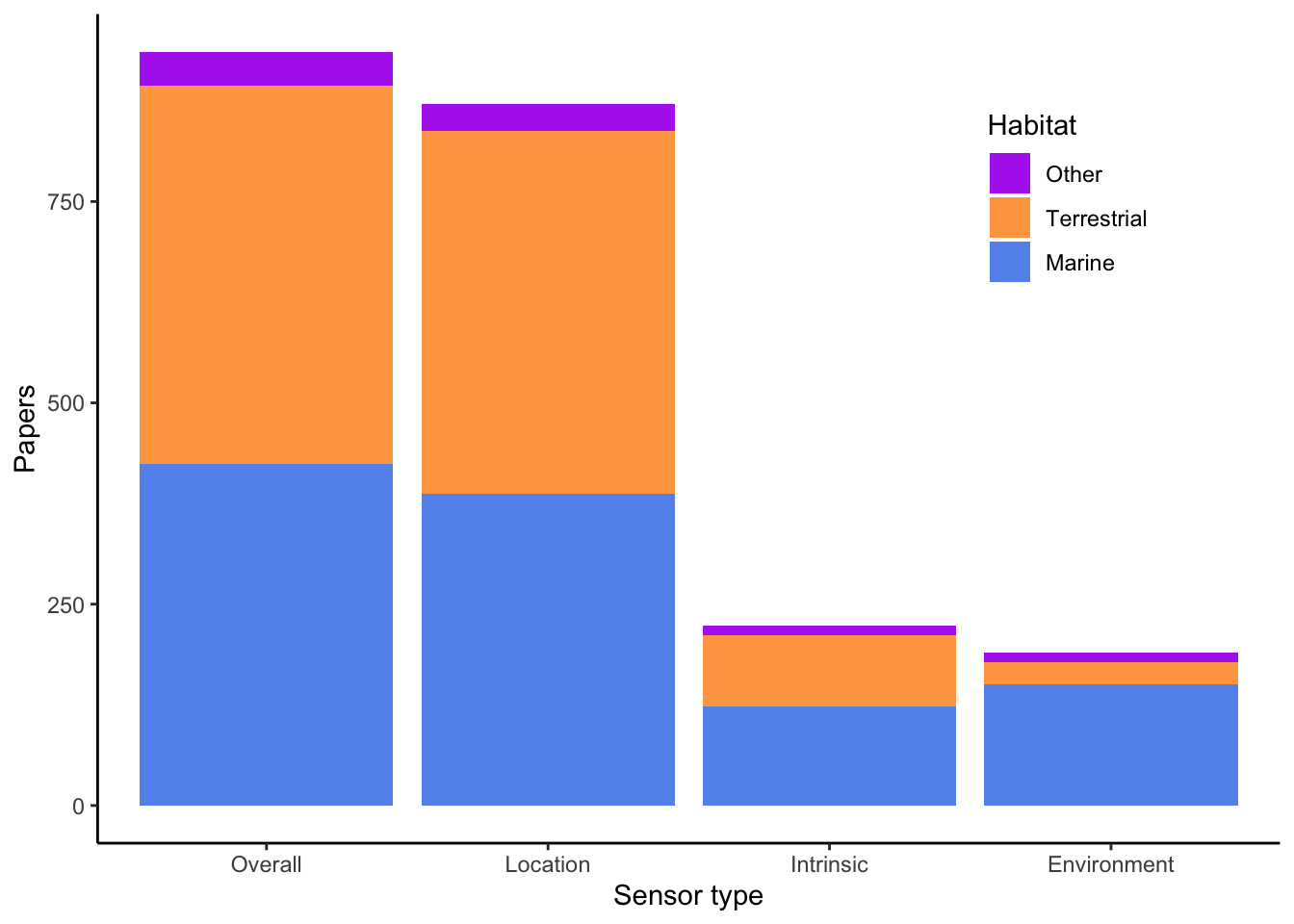
Breakdown of biologging data by sensor type. Limited to animals in the wild (no domestic or captive).
Code
<- scored_papers %>% filter (manuscript_type != "U" ,== "Y" ,str_detect (biologging_context, "W" ),! is.na (novel_biologging)) %>% mutate (Habitat = case_when (substr (habitat, 1 , 1 ) == "M" ~ "Marine" ,substr (habitat, 1 , 1 ) == "T" ~ "Terrestrial" ,TRUE ~ "Other" %>% group_by (Habitat) %>% summarize (Overall = n (),Location = sum (str_detect (device_cat, "L" ),na.rm = TRUE ),Intrinsic = sum (str_detect (device_cat, "I" ), na.rm = TRUE ),Environment = sum (str_detect (device_cat, "E" ), na.rm = TRUE )) %>% pivot_longer (- Habitat, names_to = "Sensor type" , values_to = "Papers" ) %>% mutate (` Sensor type ` = fct_reorder (` Sensor type ` , .fun = \(x) - sum (x)),Habitat = fct_reorder (Habitat, Papers, .fun = sum)%>% ggplot (aes (` Sensor type ` , Papers, fill = Habitat)) + geom_col () + scale_fill_manual (values = c (Terrestrial = "tan1" ,Marine = "cornflowerblue" ,Other = "darkorchid2" )) + theme_classic () + theme (legend.position = c (0.9 , 0.9 ),legend.justification = c (1 , 1 ))
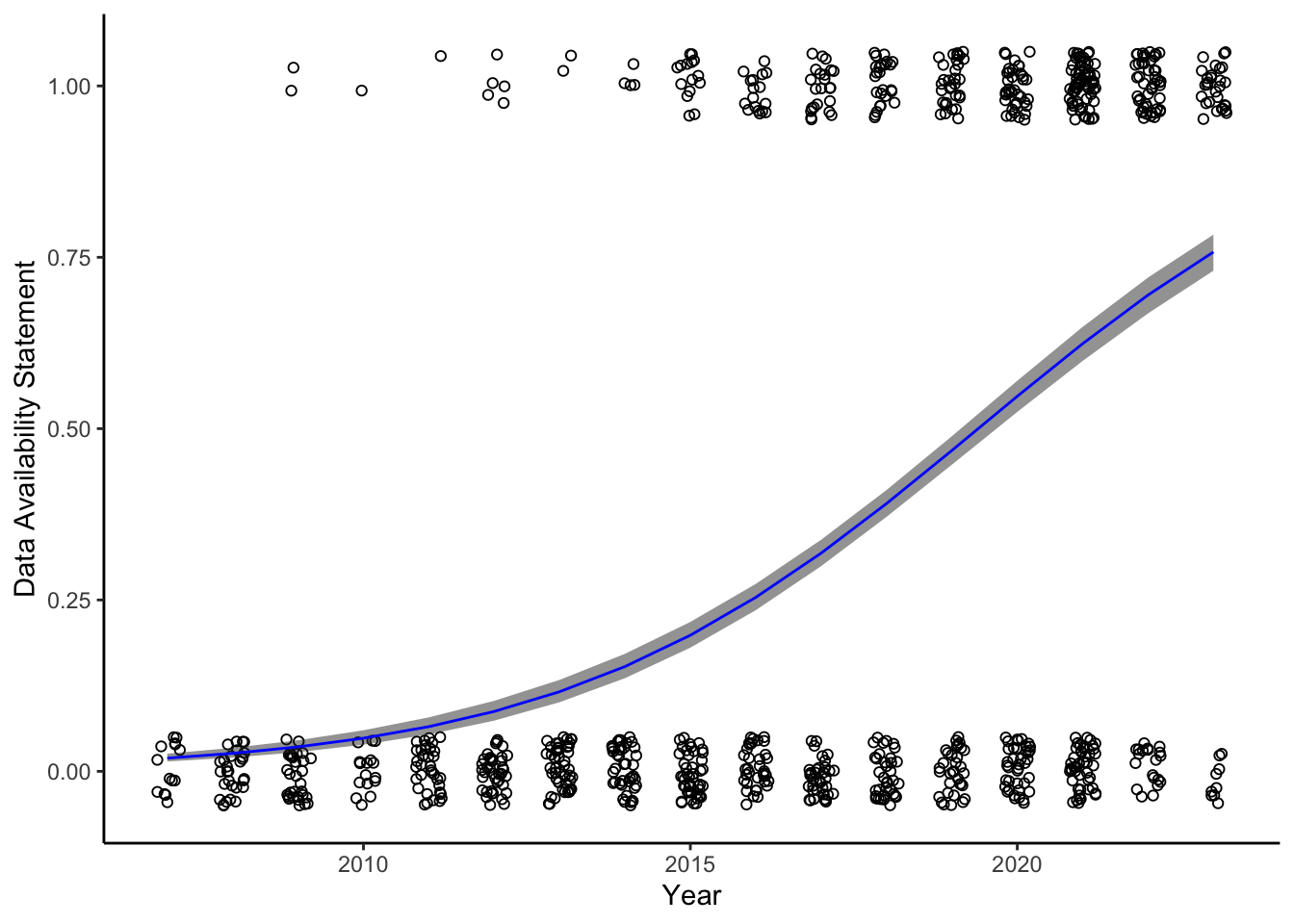
Data availability statements over time.
Code
<- novel_biolog %>% drop_na (biologging_availability) %>% mutate (biologging_availability = ifelse (biologging_availability == "Y" ,1 , 0 ),year = as.numeric (year),year2007 = year - 2007 <- glm (biologging_availability ~ year2007,family = "binomial" ,data = data_avail)<- tibble (year2007 = 0 : 16 )<- predict (data_avail_mod,newdata = data_avail_grid,se.fit = TRUE )<- binomial ()$ linkinv<- data_avail_grid %>% mutate (eta = data_avail_pred$ fit,eta_lwr = data_avail_pred$ fit - data_avail_pred$ se.fit,eta_upr = data_avail_pred$ fit + data_avail_pred$ se.fit,biologging_availability = invlogit (eta),biologging_availability_lwr = invlogit (eta_lwr),biologging_availability_upr = invlogit (eta_upr),year = year2007 + 2007 )ggplot (data_avail, aes (year, biologging_availability)) + geom_point (shape = 21 , position = position_jitter (width = 0.2 , height = 0.05 )) + geom_ribbon (aes (x = year,ymin = biologging_availability_lwr,ymax = biologging_availability_upr),alpha = 0.5 ) + geom_line (data = data_avail_pred_df,color = "blue" ) + labs (x = "Year" , y = "Data Availability Statement" ) + theme_classic ()
Taxonomic coverage of biologging.
Code
library (networkD3)<- study_taxa %>% mutate (group1 = ifelse (phylum == "Chordata" , "Vertebrate" , "Invertebrate" ),group2 = case_when (== "Aves" ~ "Birds" ,== "Mammalia" ~ "Mammals" ,%in% c ("Chondrichthyes" , "Chondrostei" , "Teleostei" ) ~ "Fish" ,== "Vertebrate" ~ "Other vertebrates" ,TRUE ~ phylumgroup3 = case_when (== "Carnivora" ~ "Carnivores" ,%in% c ("Artiodactyla" , "Perissodactyla" ) ~ "Ungulates" ,== "Cetacea" ~ "Cetaceans" ,== "Mammalia" ~ "Other mammals" ,%in% c ("Charadriiformes" , "Sphenisciformes" , "Procellariiformes" , "Suliformes" , "Pelecaniformes" ) ~ "Seabirds" ,%in% c ("Anseriformes" , "Galliformes" ) ~ "Fowl" ,%in% c ("Accipitriformes" , "Falconiformes" , "Strigiformes" ) ~ "Raptors" ,== "Aves" ~ "Other birds" ,TRUE ~ NA <- study_taxa_categorized %>% pivot_longer (group1: group3, names_to = "group" , values_to = "category" ) %>% count (category) %>% drop_na ()<- rbind (select (study_taxa_categorized, source = group1, target = group2),select (study_taxa_categorized, source = group2, target = group3)%>% drop_na (source, target) %>% count (source, target) %>% left_join (select (category_sizes, source = category, source_n = n), by = "source" ) %>% left_join (select (category_sizes, target = category, target_n = n), by = "target" ) %>% mutate (source = str_glue ("{source} ({source_n})" ),target = str_glue ("{target} ({target_n})" ))<- tibble (name = unique (c (links$ source, links$ target))$ IDsource <- match (links$ source, nodes$ name) - 1 $ IDtarget <- match (links$ target, nodes$ name) - 1 sankeyNetwork (Links = as.data.frame (links), Nodes = as.data.frame (nodes),Source = "IDsource" , Target = "IDtarget" ,Value = "n" , NodeID = "name" , sinksRight = FALSE , fontSize = 14 , fontFamily = "Helvetica" )